1. The original intention of Degpred
The ubiquitin-proteasome system (UPS) dynamically regulates protein turnover in most cellular
pathways, like cell differentiation,
cell cycle and signaling pathways, over 80% intracellular proteins are degraded by the UPS. During the
degradation process, ubiquitin was covalently attached to lysine (K) on the substrate, which is catalyzed by
E1 ubiquitin-activating enzymes, E2 ubiquitin-conjugating enzymes, and E3 ubiquitin ligases,
subsequently, the
ubiquitinated substrate is transferred to 26S proteasome and degraded. The human genome encodes 2 E1s, 41
E2s,
and more than 600 E3s. E3s bind substrates directly, and the E3 binding sites present on substrates are
named
degrons. Interactions between E3s and degrons determine the specificity of the degradation
process.
Degrons locate preferentially in disordered regions and undergo disorder-to-order
transition upon binding to E3s. Degrons are commonly regulated by post-translational modifications
(PTMs) to regulate the interaction with E3s in response to environmental and cellular cues. A key
property of degrons is their
transferability: in most cases, transplantation of a degron to a protein will accelerate the degradation of
the protein. Disfunction of degrons disturbs protein degradation and causes abnormal accumulation of
proteins, which further contributes to the progression of diseases. Thus, identification of degrons on the
substrates will help investigate the pathogenesis of diseases and provide potential therapeutic targets.
Only few bioinformatic tools have been developed to predict possible degrons. Motif
matching is widely used in the prediction of degrons, 25 degron motifs from the publicly available ELM motif
database are commonly used. While motif matching excels in predicting possible degrons fast from local
sequence patterns, it fails to consider other critical features such as structure or solvent accessibility
of specific sites, which leads to high false positive rate of motif matches. To reduce the false positive
rate of motif matching, Martínez-Jiménez, et al. matched human proteome using motifs and classified motif
matches using a random forest classifier with 11 biochemical features, including
flanking phosphorylation sites, intrinsically disordered regions, MoRFs, solvent accessibility and flanking
ubiquitin Ks. That study identified over 20,000 likely new degrons in different protein isoforms.
Nevertheless, both methods use less than 30 E3 motifs and only cover degrons for less than 5% E3s compared
to more than 600 E3s, precluding us from identifying degrons bound by other E3s. Tokheim, et al. trained a
deep learning model to predict N-end and C-end degrons using high-throughput GPS data11. However, this
method can only predict terminus located degrons and its training set from GPS experiment not only includes
degrons, thus the prediction result is a mixture of multiple destabilizing peptides. Thus, a more general
model is needed to give a broader and unbiased prediction of degrons.
Besides identification of degrons, identifying E3-substrate interactions (ESIs) is also an area
of intense study. Co-immunoprecipitation, two-hybrid screen and mass spectrometry-based methods are commonly
used to experimentally discover new ESIs. Recently, Machine learning was applied to predict ESIs as well.
Wang et al. developed Ubibrowser 2.0 to predict ESIs using enriched domain, GO term pair, protein–protein
interaction and inferred E3 recognition consensus motif; Chen et al. built a machine learning model
to predict ESIs from proteomics data, transcriptomics data, protein–protein interaction
and pathway-based associations. However, both experimental methods and prediction methods lack binding
degron information.
2. Degron collection and Degpred construction
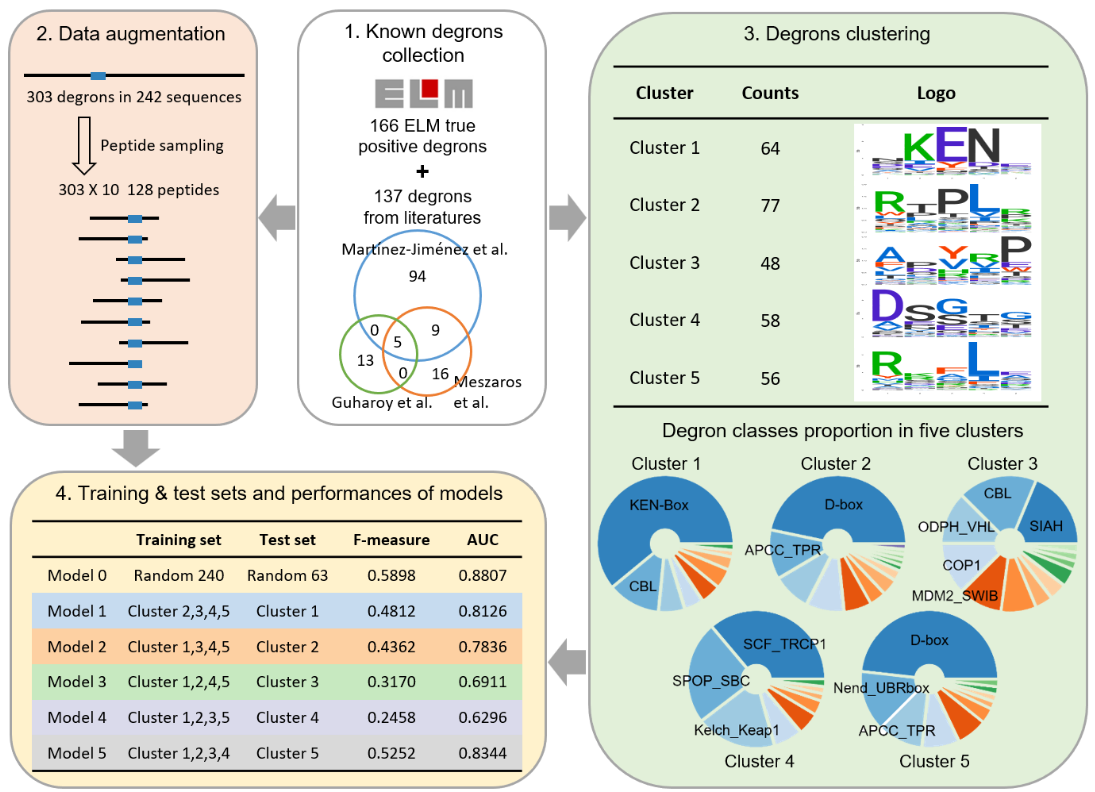
1) Data collection:
Known degrons were collected from ELM database and previous studies. For the same degrons on
different isoforms
of one gene, only the main isoforms in UniProt were reserved. In total, 303 degrons typically spanned 5–10
residues were obtained. But the repertoire of known degrons is still very limited, thus, we augmented these
degrons by peptides sampling. For each degron, ten peptides of 128 AAs containing the degron were randomly
sampled from the protein, thus, we got 3030 128AA-peptides. According to the transferability of degrons,
degrons on
128AA-peptides can mediate the degradation of 128-peptide as well.
2) Model construction:
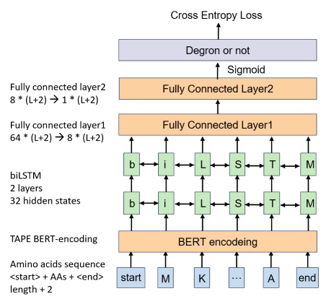
We grouped known degrons into 5 clusters by sequence alignment. Cluster 1, 2, 4, 5 possessed dominant classes that
accounted for about half of clusters, while cluster 3 had no major class and acted as a trash bin during
clustering.
Five models were built and each model was trained on four clusters and tested on the cluster
left. Model 1, 2, 5
achieved promising performances, considering diverse degrons in cluster 3, the performance of Model 3 was
also
satisfactory. For Model 4, the dominant class was a kind of phospho-degron, and phosphorylation is necessary
for
them to be bound by βTrCP. As degrons used to train Model 4 are mostly modification independent, Model 4
might
ignore features about PTMs and performed relatively poor in predicting phospho-degrons. However,
phospho-degrons
can be predicted by the other four models as cluster 4 was included in their training set. To take full
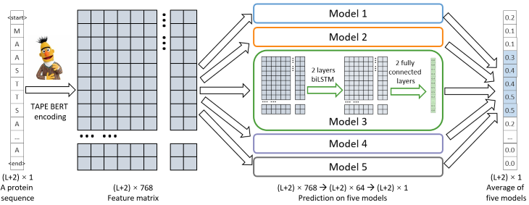
advantage of known degrons and provide comprehensive prediction, we assembled Model 1-5 to build Degpred,
which took the average of outputs from 5 models to provide scores for all AAs of the input protein.
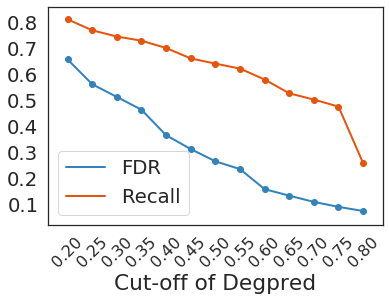
Taking 0.3 as the cutoff, Degpred attained an FDR of 0.512 at positive: negative = 1:20, negative samples were AAs randomly selected from proteins possessing test degrons. We predicted 46,621 degrons in UniProt human reviewed proteome, and only 5,522 predicted degrons overlapped with ELM motif matches. Even though most of training degrons came from ELM motif matches, over 88% predicted degrons can’t be discovered using motifs.
3. Assigning predicted degrons to E3s using calculated motifs
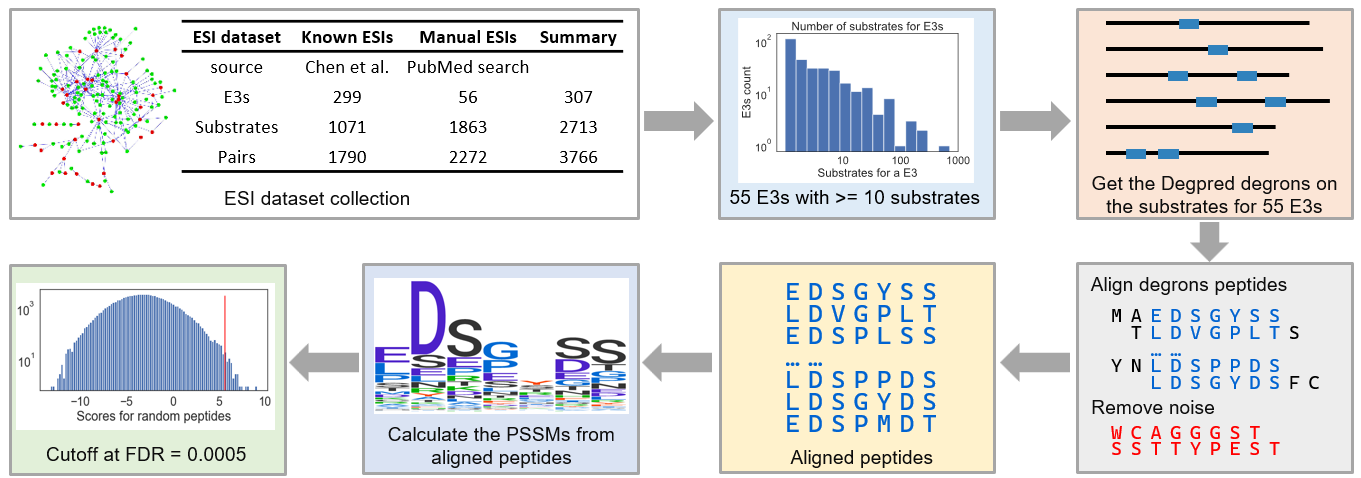
We collected an ESI dataset from PubMed and related databases. 965 related papers were obtained by searching PubMed with keywords:
(E3[Title] OR E3s[Title]) AND (substrate[Title/Abstract] OR substrates[Title/Abstract])
27 of them were retained after manual screening. 2272 nonredundant ESIs
between 56 human E3s and 1863 substrates were extracted from these studies, most of them came from high
throughput experiments. In addition, Chen et al. had collected 1790 nonredundant known ESIs from BioGrid,
E3Net, hUbiquitome and UniProt, these ESIs were included as well. In total, we got 3766 nonredundant ESIs
between 307 human E3s and 2713 substrates.
Fifty-five E3s with more than 10 substrates were chosen for motif calculation. For each E3, we
used
GibbsCluster to align predicted degrons on its substrates and drop the outliers which can’t match well with
other degrons, actually, the dropped degrons may be the binding sites for other E3s. Finally, motifs were
calculated from the aligned Degpred degrons.
Next, we calculated cutoffs for motifs and defined that if the matching score between a
predicted degron and
a motif surpasses the cutoff of the motif, the degron is considered as the binding site of the E3 and the
protein is considered as a substrate of the E3. Then, we evaluated the ability of these motifs in predicting
known ESIs and selected 39 motifs that can capture no less than 40% known substrates for further analyses.
Finally, we predicted 25695 ESIs between 39 E3s and 8754 substrates.